

Proteomics analysis
Introduction:
Proteomics is a high throughput technology for display, identify and characterization of all or a subset of proteins in a single cell, tissue or organism. The ultimate goal of proteomics research is to characterize the status of all proteins in a biological system. Depending on the depth of research, proteomics techniques can be assigned into the following levels:
1、Protein identification and protein sequence analysis: to identify the protein and protein amino sequence with mass spectrometry and other technologies.
2、Protein quantification (abundance analysis): relative quantification or absolute quantification of proteins in one sample (the amount of a certain protein in the sample) or different samples (wild type vs. mutant, treatment vs. control).
3、PTM analysis: identify and validate protein post-translational modification status.
4、Protein-protein interaction: identify protein-protein interaction by Co-IP and chemical cross-linking coupled with mass spectrometry.
There are two commonly used approaches to identify and characterize proteins using MS: “bottom-up” and “top-down”. Top-down proteomics approach obtains mass-to-charge ratio and molecular weight of intact proteins. Collision induced fragmentation of protein is used to analyze protein sequence. The major advantages of the top-down strategy are the potential to access complete protein sequence and the capability to locate and characterize PTMs. The approach requires extended scan range, sensitivity, and resolution of the mass spectrometer when analyzing larger proteins. In most cases, super-resolution orbitrap mass spectrometer and FT-ICR mass spectrometer are the instrument of choice for top-down approach.
Bottom-up proteomics involves proteolytic digestion of proteins before mass spectrometric analysis. The resulted peptides are analyzed by mass spectrometry. Proteins are identified from the acquired mass spectra using database searching software. Bottom-up proteomics is also called shot-gun proteomics as digestion from protein to peptides is similar to the process of shooting the protein into pieces with a shot-gun. The bottom-up workflow solved many problems that come with the top-down approach, such as extraction of in-soluble membrane proteins and limited scan range and resolution of the mass spectrometers. The digested peptides usually have 7-30 amino acids with a molecular weight less than 3 kDa, which is very suitable for mass spectrometric detection. Protein is identified by unique peptide sequences, instead of full sequences. Bottom-up proteomics is the most mature and most widely used proteomics approach.
The workflow of bottom-up proteomics is as follows:
1、Protein extraction: extract proteins from cells, tissues, and body fluids with lysis buffer or urea. It is optional to precipitate proteins with acetone to remove contamination. Target proteins can be enriched with Co-IP and separated with SDS-PAGE.
2、Protein digestion: proteins are digested with site-specific endoprotease, such as trypsin, Lys-C, Asp-N, chymotrypsin, and so on.
3、LC-MS analysis: peptides are separated with nano-HPLC (hydrophilic peptides elute early and hydrophobic peptides elute later). Peptides and their fragment ions are analyzed with high-resolution mass spectrometer.
4、Data analysis: database searching software is used for protein identification and quantification.
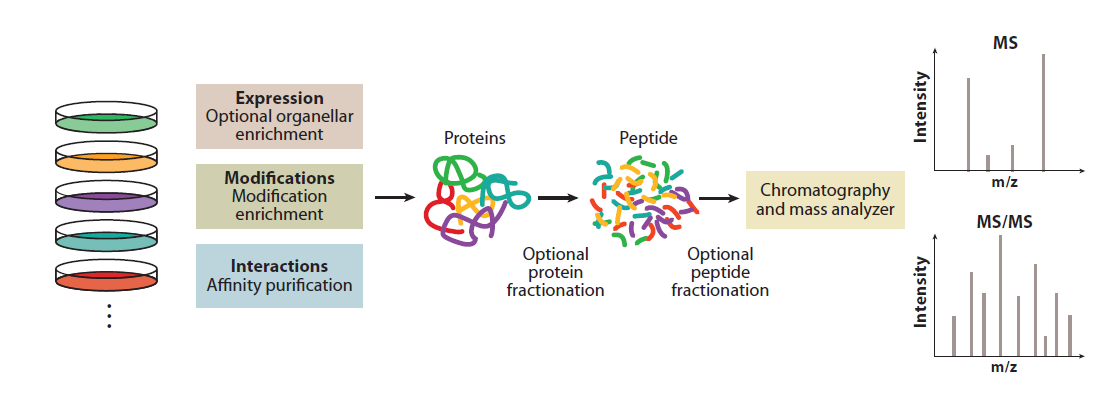
Schematic illustration of proteomics analysis
Cox & Mann, Annu. Rev. Biochem. 2011 80 273
Simple proteomics analysis can identify and verify target protein bands in SDS-PAGE, identify protein-protein interaction of Co-IP experiments, and locate protein post-translational modification sites. Advanced proteomics experiments performs large scale analysis of hundreds, even thousands of samples and quantifies more than 10,000 types of proteins, it can uncover differentially-expressed proteins under different diseases or physiological states to investigate the mechanism of diseases and drug treatments.
Sample submission:
For in-gel samples, the bands should be visible with Coomassie Blue staining or silver staining. For solution samples, sample information such as protein concentration and buffer composition should be informed. Must not contain surfactants such as SDS, RIPA, CHAPS, and Triton. For quantitative proteomics experiments, please provide at least 1×107 cells or at least 10 mg of tissue. For protein modification analysis, if enrichment is necessary, please inform BMCS in advance. If endoprotease other than trypsin is needed, please inform BMCS in advance.
Result output:
The results will be sent by email. If you need the original raw data, please download from data storage server at HPC core facility.

Address :Chinese Institute for Brain Research, Beijing(CIBR), Jianzan Building, No.26, Science Park Road, Zhongguancun Life Science Park, Changping District, Beijing, China
-
Contact:010-80765986 Postalcode:102206
© 2023 by Personal Life Coach. Proudly created with Wix.com ICP备案号:京ICP备18029179号